Research
‘Research is to see what everybody else has seen, and to think what nobody else has thought.’
Albert Szent-Gyorgyi
Olefin Metathesis
The creation of C-C double bonds under simultaneous redistribution of alkene fragments is one of the most important catalytic reaction in the chemical industry being used to manifacture polymers, to synthesize new drugs or to convert biomass.
Essential for the success is the development of highly active catalysts. We investigate the reactivity and selectivity in olefin metathesis with a novel class of Mo imido alkylidene N-heterocyclic carbene complexes that show high productivity and functional group tolerance in the monomers. Our ultimate goal is to suggest superior catalysts.
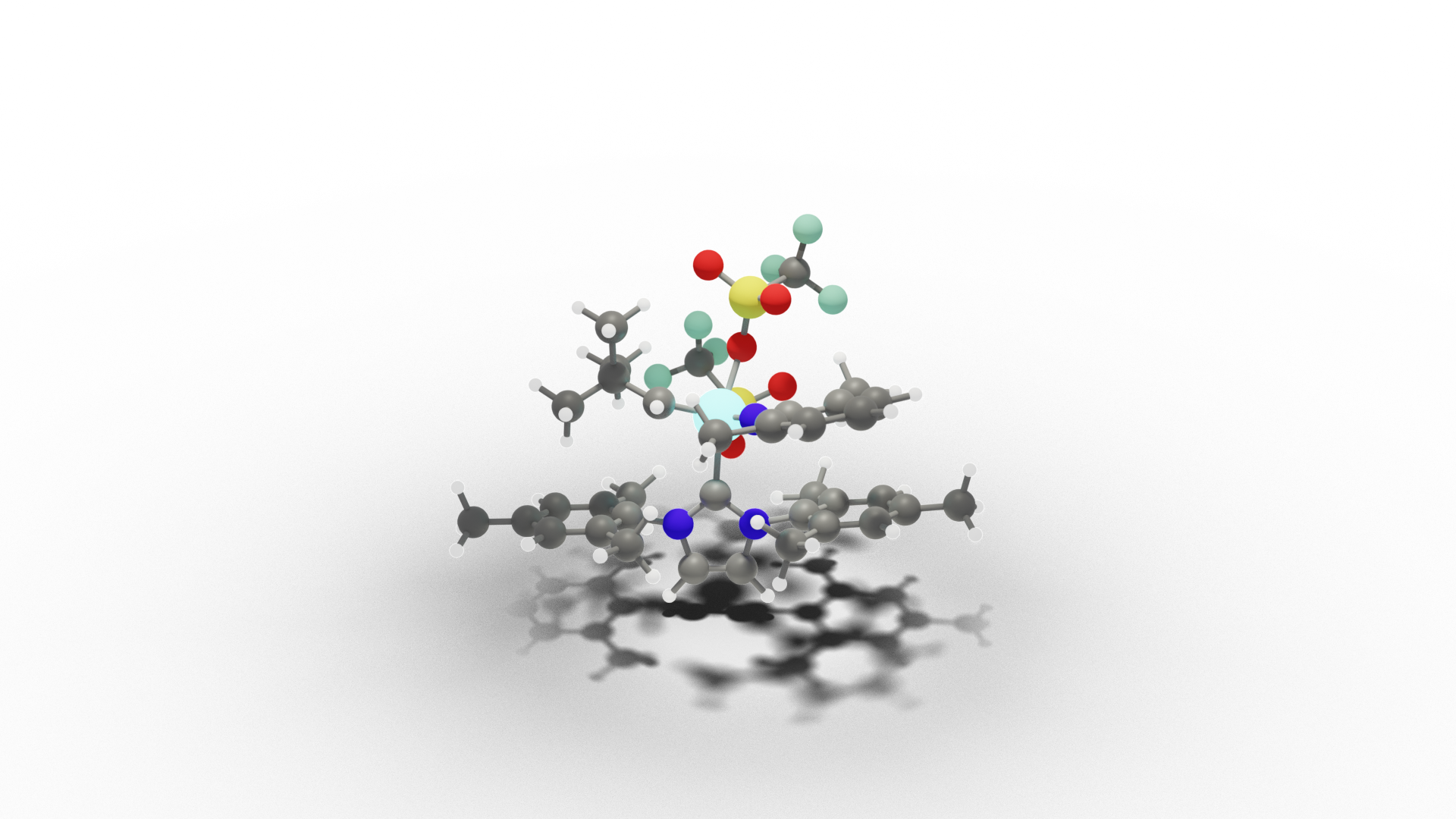
Catalysis Under Confinment
Catalysis under confinement is a concept Nature has perfected in enzymes, where the binding pockets provide an ideal environment to achieve a chemical transformation at ambient conditions with utmost efficiency. This concept has inspired researchers to develop artificial cavities to enhance the performence of the catalyst and to mimic nature. In homogeneous catalysis, macrocyclic ligands have emerged as a powerful tool to encapsulate transition metals to emulate the behavior of metalloenzymes. Yet little is known about the origins of these catalytic enhancements. Our goal is to develop computational multilevel protocols that capture the flexibility of the cavity. Out ultimate aim is to study the reactivity and mechanism of such supramolecular catalysts.

Quantum Chemical Microsolvation
Well-known in experimental chemistry, the choice of an appropriate solvent impacts the outcome of a reaction by influencing selectivity, reactivity and stability of the reactants and intermediates. Yet, systematic quantum chemical studies on complex solvent effects are scarce due to the difficulty to model explict solute-solvent effects in a quantum chemical framework. The high computational costs of ab initio condensed phase approaches limit its applicability to small systems only. A low cost alternative are microsolvation approaches, where only a few solvent molecules are explicitly described. We developed a methodology to automatically select and place individual solvent molecules based on thermodynamic properties as starting structures for subsequent quantum chemical investigations.

Methodology
To address the outlined research questions, an accurate description of chemical system is crucial. Therefore, we use a broad range of computational methods ranging from molecular dynamics and enhanced sampling techniques to (ab initio) electronic structure theory. Our goal is to develop tailored multi-level protocols suited to describe the structure and dynamics of the system under study.
